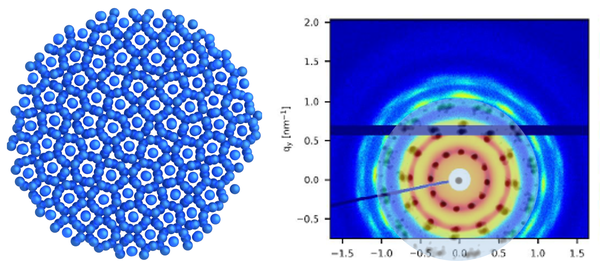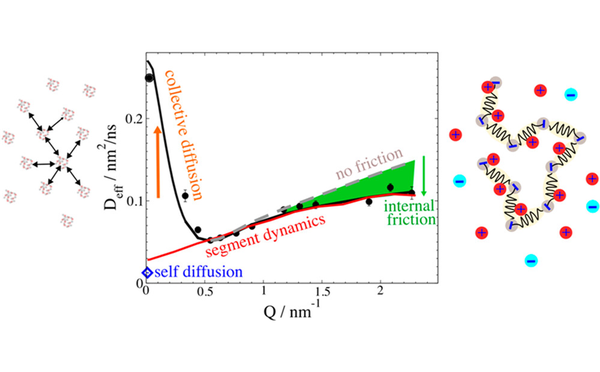
Inter- und Intrapartikeldynamik von Makromolekülen
Durch die Kombination von dynamischer Lichtstreuung, NMR und Neutronenspinecho ist es uns möglich die dynamischen Prozesse inter als auch intramolekular von Proteinen, Polymeren und Polyelectrolyten zu untersuchen.