Atomistic Simulations of Crystal Defects
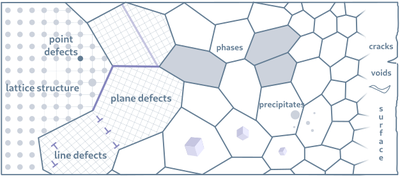
The physical properties of materials are profoundly influenced by defects in their crystal structures. Our research employs computational materials science, including ab initio and atomistic simulations, to investigate these defect structures. From the insights at the atomic scale, we aim to address challenges in materials design and derive structure-property relationships. A key focus of our work is the development of a crystal defects knowledge graph, enabling efficient exploration of data spaces that connect atomic structures, material properties, and thermodynamic states.
Contact:
Dr. Abril Azocar Guzman
Tel.: +49 241/927803-43
E-mail: a.azocar.guzman@fz-juelich.de
Last Modified: 22.10.2025