Transportprozesse in gesunden und pathologisch veränderten Zellen
Zellen benutzen Ionenkanäle und –transporter, um elektrische Signale zu generieren, weiterzuleiten und auf andere Zellen zu übertragen. Da das Lipidgrundgerüst der Zellmembran für alle geladenen und polaren Substanzen undurchlässig ist, sind Ionenkanäle und –transporter für den Transport von Nährstoffen, Elektrolyten und Signalmolekülen in Zellen hinein oder aus ihnen heraus von entscheidender Bedeutung.
Verschiedene menschliche Erkrankungen werden durch genetische und erworbene Veränderungen von Ionenkanälen und –transportern verursacht. Die Untersuchung der Pathophysiologie dieser Erkrankungen erlaubt wichtige Einblicke in zelluläre Aufgaben dieser Proteine. Da Ionenkanäle und –transporter attraktive Ziele für pharmakologische Modifikation sind, versprechen derartige Untersuchungen auch die Entwicklungen neuer therapeutischer Ansätze.
Wir untersuchen die molekulare Physiologie und Pathophysiologie von Transportvorgängen. Unser Ziel ist es, die Mechanismen von Membrantransportprozessen zu verstehen, mögliche Ursachen ihrer Dysfunktionen aufzudecken und die Auswirkungen von gestörten Transportvorgängen auf die Organfunktion zu beschreiben. Wir versuchen dabei, den Bogen von molekularen Vorgängen in einzelnen Transportproteinen über ihre zellulären Funktionen bis hin zu Organfunktionen zu spannen.
Wir verwenden dazu eine Vielzahl verschiedener Methoden. Wir benutzen elektrophysiologische Verfahren, wie beispielsweise patch-clamp-Messungen oder Mikroelektrodenableitungen, Fluoreszenz-Indikatormessungen und radioaktive Methoden, um Transportfunktionen direkt zu beobachten. Wir haben angefangen, mit computer-gestützten Verfahren Transportprozesse auf atomarer Ebene zu simulieren und zu verstehen. Um Sequenz-Determinanten von Transportprozessen zu identifizieren, werden Kanäle und Transporter - mit normaler oder gerichtet veränderter Proteinsequenz - heterolog in Zellen und Xenopus Oozyten exprimiert. Wir exprimieren Proteine in Bakterien, reinigen sie auf und untersuchen sie mit fluoreszenzspektroskopischen Methoden. Wir benutzen Ableitungen an dissoziierten Neuronen und Gliazellen sowie Gehirnschnittpräparationen, um die Funktion von Transportproteinen in nativen Geweben zu charakterisieren. Um Auswirkungen von krankheitsverursachenden Mutationen auf bestimmte Organfunktionen zu untersuchen, benutzen wir darüber hinaus Mausmodelle.
Wir konzentrieren uns zurzeit auf drei Klassen von Membranproteinen, auf ClC Anionenkanäle und Transporter, auf die SLC26 Klasse multifunktioneller Anionentransporter und auf Glutamattransporter. Die Bedeutung dieser Proteinklassen wird nachdrücklich durch die Existenz monogenetischer Erkrankungen unterstrichen, die durch die Dysfunktion dieser Transporter verursacht wird.
Glutamattransporter in episodischer Ataxie und Epilepsie
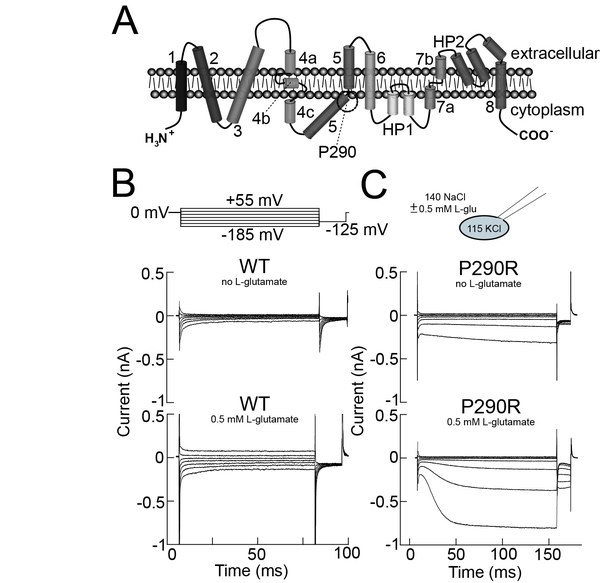
Episodische Ataxien sind seltene menschliche Erkrankungen, bei denen es anfallsweise zu Störungen der Bewegungskoordination kommt. Eine Form der episodischen Ataxie wird durch Mutationen in dem SLC1A3 Gen, das für den glialen Glutamattransporter EAAT1 kodiert, verursacht. EAAT Glutamattransporter weisen die besondere Eigenschaft auf, dass sie neben sekundär-aktivem Glutamattransport außerdem noch als Anionenkanäle funktionieren können. Wir beschäftigen uns mit einer krankheitsverursachenden Mutation, die den Austausch eines hochkonservierten Prolin an Position 290 durch Arginin verursacht. Diese Mutation wurde in einem Patienten beschrieben, der neben Ataxien auch unter einer ausgeprägten Epilepsie litt.
P290R reduziert den Glutamattransport, erhöht den EAAT1 Anionenstrom aber dramatisch. Wir untersuchen die molekularen Mechanismen der P290R-vermittelten Änderung beider EAAT Transportfunktionen. Um zu verstehen, wie die Veränderung einer glialen Anionenleitfähigkeit zur neuronalen Übererregbarkeit führen kann, haben wir ein Mausmodell generieren lassen, das die krankheitsverursachende Mutation trägt und das wir zurzeit mit elektrophysiologischen und mikroskopischen Verfahren untersuchen. P290R knock in Mäuse weisen eine ausgeprägte Ataxie und Epilepsie auf.
Ionenkanäle in der Blutdruck-Regulation
Die Aussscheidung von Elektrolyten und Wasser durch die Niere ist ein entscheidender Regulator des Blutdrucks. Veränderungen in der Funktion von Ionenkanälen im Nephron oder in endokrinen Zellen, die Hormone freisetzen die die Nierenfunktion modifizieren, sind mögliche Ursachen des Bluthochdrucks.
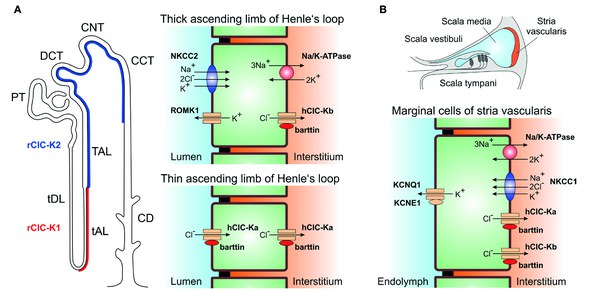
Anionenkanäle im dünnen und dicken aufsteigenden Teil der Henle Schleife sind für die Harnkonzentration von großer Bedeutung. Sie bestehen aus zwei porenbildenden ClC-K Untereinheiten und einer bislang unbekannten Anzahl der akzessorischen Untereinheit Barttin. ClC-K Kanäle sind nur dann funktionell, wenn sie zusammen mit Barttin exprimiert werden. Mutationen in den Genen für ClC-Ka (CLCNKA) und ClC-Kb (CLCNKB) sowie für Barttin (BSND) verursachen Störungen in der Blutdurckregulation und können zu erblicher Schwerhörigkeit führen. Wir untersuchen durch eine Kombination aus Molekularbiologie, Elektrophysiologie, „super-resolution microscopy“ und computer-gestützten Verfahren, wie Barttin ClC-K Kanäle reguliert. Wir hoffen mit diesen Arbeiten neue pharmakologische Ansätze definieren zu können, um diese Kanäle spezifisch zu blockieren.
Transportprozesse in synaptischen Vesikeln
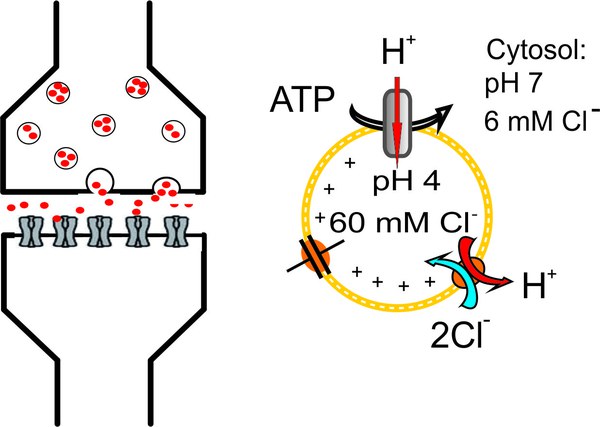
Neuronen können über chemische Synapsen miteinander kommunizieren. Chemische Synapsen passen sich in ihrer Funktion der synaptischen Aktivität an und sind daher für komplexe zentralnervöse Prozesse von besonderer Bedeutung.
Die synaptische Übertragung beginnt mit der Freisetzung eines Neurotransmitters durch die Fusion synaptischer Vesikel mit der oberflächlichen Membran der präsynaptischen Nervenendigung. Wir untersuchen, wie Neurotransmitter in synaptischen Vesikeln angereichert werden und welche besonderen Transportprozesse dafür notwendig sind. .
Publikationen zum Thema
Ewers, D., Becher, T., Machtens, J. P., Weyand, I., and Fahlke, Ch. (2013) Induced fit substrate binding to an archeal glutamate transporter homologue. Proc Natl Acad Sci U S A 110, 12486-12491
Fahlke, Ch., Rüdel, R., Mitrovic, N., Zhou, M., and George, A. L., Jr. (1995) An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron 15, 463-472
Fahlke, Ch., Beck, C. L., and George, A. L., Jr. (1997) A mutation in autosomal dominant myotonia congenita affects pore properties of the muscle chloride channel. Proc Natl Acad Sci USA 94, 2729-2734
Fahlke, Ch., Yu, H. T., Beck, C. L., Rhodes, T. H., and George, A. L., Jr. (1997) Pore-forming segments in voltage-gated chloride channels. Nature 390, 529-532
Fischer, M., Janssen, A. G., and Fahlke, Ch. (2010) Barttin activates ClC-K channel function by modulating gating. J Am.Soc.Nephrol. 21, 1281-1289
Gendreau, S., Voswinkel, S., Torres-Salazar, D., Lang, N., Heidtmann, H., Detro-Dassen, S., Schmalzing, G., Hidalgo, P., and Fahlke, Ch. (2004) A trimeric quaternary structure is conserved in bacterial and human glutamate transporters. J Biol.Chem.279, 39505-39512
Melzer, N., Biela, A., and Fahlke, Ch. (2003) Glutamate modifies ion conduction and voltage-dependent gating of excitatory amino acid transporter-associated anion channels. J Biol.Chem. 278, 50112-50119
Riazuddin, S., Anwar, S., Fischer, M., Ahmed, Z. M., Khan, S. Y., Janssen, A. G., Zafar, A. U., Scholl, U., Husnain, T., Belyantseva, I. A., Friedman, P. L., Riazuddin, S., Friedman, T. B., and Fahlke, Ch. (2009) Molecular basis of DFNB73: mutations of BSND can cause nonsyndromic deafness or Bartter syndrome. Am.J.Hum.Genet. 85, 273-280
Scholl, U. I., Goh, G., Stolting, G., de Oliveira, R. C., Choi, M., Overton, J. D., Fonseca, A. L., Korah, R., Starker, L. F., Kunstman, J. W., Prasad, M. L., Hartung, E. A., Mauras, N., Benson, M. R., Brady, T., Shapiro, J. R., Loring, E., Nelson-Williams, C., Libutti, S. K., Mane, S., Hellman, P., Westin, G., Akerstrom, G., Bjorklund, P., Carling, T., Fahlke, Ch., Hidalgo, P., and Lifton, R. P. (2013) Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet45, 1050-1054
Scholl, U., Hebeisen, S., Janssen, A. G., Muller-Newen, G., Alekov, A., and Fahlke, Ch. (2006) Barttin modulates trafficking and function of ClC-K channels. Proc.Natl.Acad.Sci.U.S.A 103, 11411-11416
Stölting, G., Teodorescu, G., Begemann, B., Schubert, J., Nabbout, R., Toliat, M. R., Sander, T., Nurnberg, P., Lerche, H., and Fahlke, Ch. (2013) Regulation of ClC-2 gating by intracellular ATP. Pflugers Arch 465, 1423-1437
Winter, N., Kovermann, P., and Fahlke, Ch. (2012) A point mutation associated with episodic ataxia 6 increases glutamate transporter anion currents. Brain 135, 3416-3425