Atomistic Simulation of Interfaces and Surfaces

A detailed knowledge of the atomistic structure at interfaces, surfaces, multiphase boundaries, and defects is crucial for an understanding of the method of operation, the activity, the ageing, and finally the failure of both batteries and electrocatalysts. However, these areas are notoriously difficult to study both experimentally and theoretically. Only by combining various simulation methods with experiments, we stand a chance to get an insight into those challenging, clandestine zones of the highly complex systems for energy storage and conversion.
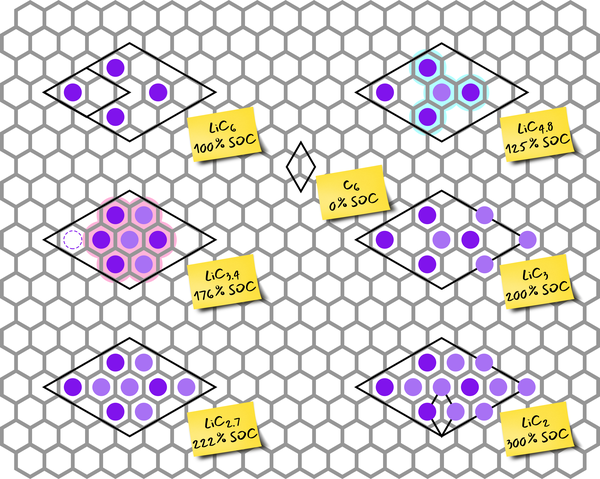
We utilize different theoretical approaches from first principle methods via molecular dynamics to kinetic Monte Carlo in a multiscale approach in order to bridge several length and time scales in a consistent way. The latest developments in data science such as machine learning are applied to increase efficiency and accessible length and time scales. Hence, a valid theoretical picture of the processes in those regions is generated, which is then correlated to experimental results in order to verify the model.
Contact

Last Modified: 17.02.2025