Atomistische Simulation von Grenz- und Oberflächen

Ein detaillierter Einblick in die atomistische Struktur an Grenzflächen, Oberflächen, Mehrphasengrenzen und Defekten ist entscheidend, um die Funktionsweise, die Aktivität, die Alterung und letztendlich den Ausfall von Batterien und Elektrokatalysatoren zu verstehen. Allerdings sind diese Bereiche notorisch schwierig zu untersuchen, sowohl experimentell als auch theoretisch. Nur durch die Kombination unterschiedlicher Simulationsmethoden mit Experimenten haben wir eine Chance, einen Einblick in diese anspruchsvollen, verborgenen Zonen hoch komplexer Systeme zur Energiespeicherung und -umwandlung zu gewinnen.
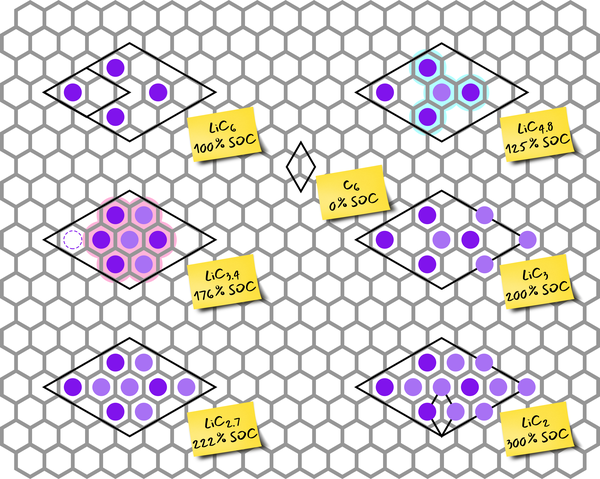
Wir nutzen verschiedene theoretische Techniken von first principles Methoden über Moleküldynamik bis hin zu kinetischer Monte Carlo in einem multiskaligen Ansatz, um mehrere Längen- und Zeitskalen auf konsistente Art und Weise zu überbrücken. Die neuesten Entwicklungen im Bereich Datenwissenschaften wie maschinelles Lernen werden eingesetzt, um die Effizienz und die zugänglichen Längen- und Zeitskalen zu steigern. Auf diese Weise wird ein fundiertes theoretisches Bild entwickelt, das die Prozesse in diesen Regionen wiedergibt. Eine anschließende Korrelation des theoretischen Modells mit experimentellen Ergebnissen dient zur Verifizierung.
Ansprechpartner

Letzte Änderung: 17.02.2025