Prion diseases
Prion diseases or transmissible spongiform encephalopathies (TSEs) are a special group of neurodegenerative diseases. They occur in humans - Creutzfeldt-Jakob disease (CJD for "Creutzfeldt-Jabob disease") and in animals - including "scrapie" in sheep, "bovine spongiform encepahlophathy" (BSE) in cattle. Prion diseases can have a spontaneous, genetic or infectious background. Transmissibility by infection distinguishes prion diseases from other neurodegenerative ones, such as Alzheimer's dementia or Huntington's disease. In the course of the disease, behavioral changes, coordinative dysfunctions (ataxia) and dementia occur neuropathologically, spongiform changes in the brain, degeneration of neurons and astrocytosis are detectable as histopathological hallmarks of prion diseases (Fig. 1).
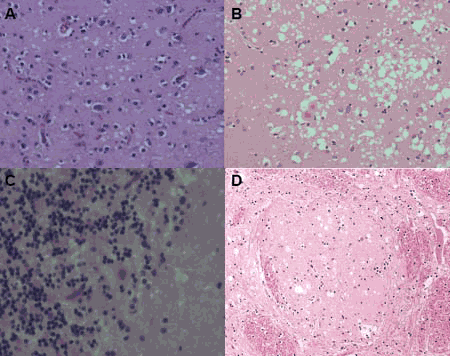
In 1982, Stanley B. Pruisiner published the prion hypothesis, which states that the causative agent of prion diseases consisted exclusively of proteinaceous particles (prions). The main component of prions is the prion protein in a misfolded structure (PrPSc). The prion protein is an endogenous protein (PrPC). The replication mechanism is based on the fact that PrPSc is able to interact with PrPC and convert PrPC into PrPSc.
We are developing a method for early diagnosis of prion disease using aggregation state as a biomarker.
In addition, we are investigating the underlying replication mechanism in vitro.