Enhanced sampling
A massively parallel algorithm for thermodynamics and kinetics of complex systems
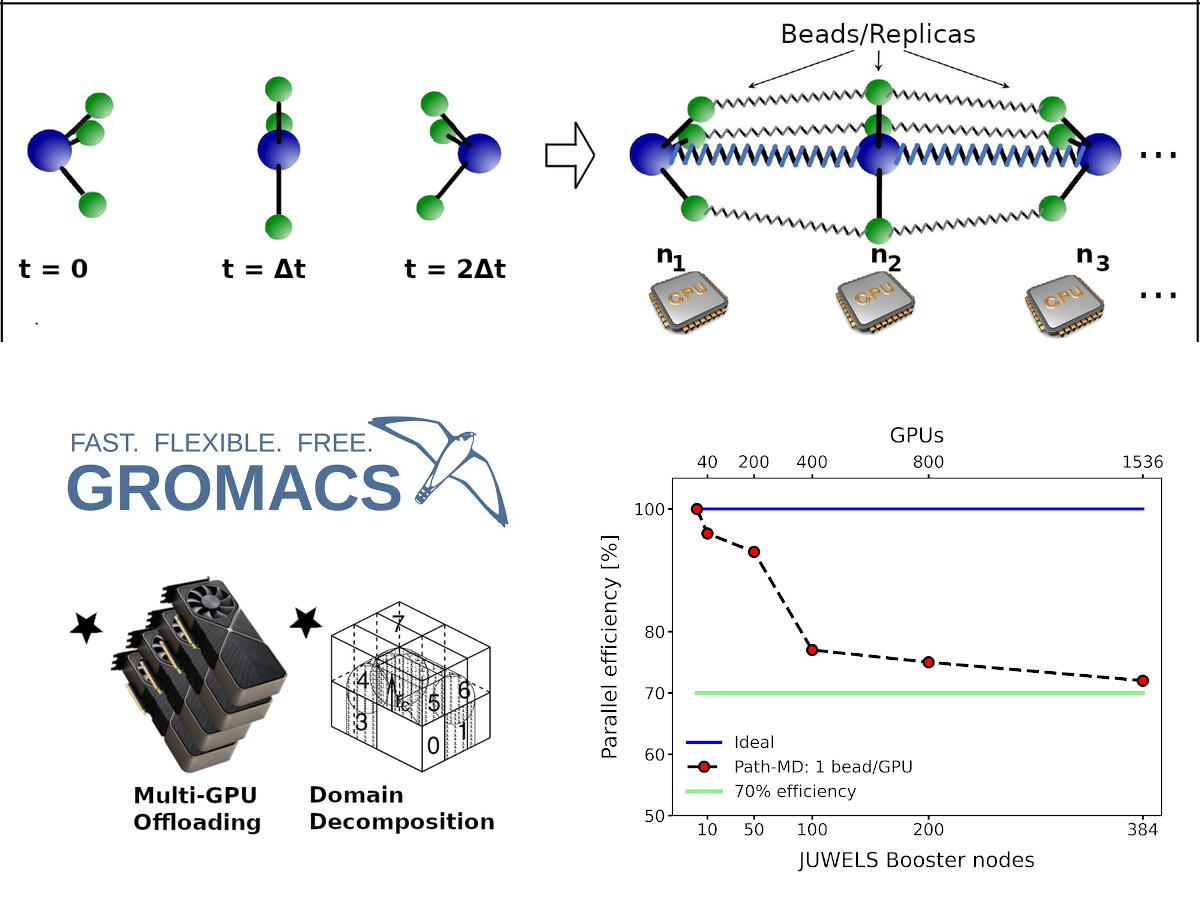
Figure 1 - (Top panel) The algorithm exploits the isomorphism between the probability of observing a trajectory of N steps and the partition function of a fictitious polymer made of N beads. The beads correspond to the configurations visited by the system along the trajectory and they interact via springs. The isomorphism allows performing MD simulations of the fictitious polymer to sample discretized physical trajectories directly from the ensemble of all possible paths. Because it deals with a system that is N times larger, the algorithm can be implemented in a highly parallel fashion, in principle allowing one to sample arbitrarily long trajectories by scaling up to any arbitrary number of computer nodes. (Bottom panel) We have implemented the algorithm in GROMACS-2024 with the possibility to exploit its main parallelization strategies, including Domain Decomposition, GPU-offloading of the force computation as well as combination of the two. Preliminary benchmarks on a G-protein coupled receptor (150 katoms) system demonstrated weak scaling parallel efficiency above 70% running using up to 1536 GPUs (384 nodes) on the pre-exascale JUWELS Booster at the Juelich Supercomputing Center.
The human brain operates at different time scales and sizes. Shedding light on processes at the molecular scale is essential to understand neurotransmission, neuronal function and dysfunction. This requires a detailed prediction of the free energy landscape associated with a variety of complex processes, such as chemo transmitter/protein-protein interactions, receptor activation, and ion transport. In some cases, these involve quantum mechanical phenomena such as bond-breaking and forming in enzymatic reactions. Classical and hybrid quantum/classical (QM/MM) atomistic molecular dynamics-based approaches can be of great help in such prediction, yet, for high complex systems such as the biomolecular complexes present at the human synapses, they still suffer from limited scalability of the existing codes. Exploiting modern GPU-based massively-parallel computer architectures is therefore crucial to improve the statistical accuracy of the predictions for these large systems.
To address these key issues, we are developing strategies building on a recently proposed HPC-based approach, called metadynamics of paths [1]. The method is based on the statistical mechanics of paths and it maps the time scale problem that limits the statistical accuracy of current approaches into a size problem in an auxiliary, enlarged configurational space (see Figure 1). This allows leveraging on massively parallel computer architectures to tackle the time scale problem to explore the conformational ensemble of complex biomolecules and compute kinetic rates of important biomolecular processes, including phrmacologically relevant phenomena like ligands (un)binding from their protein targets.
A PhD student funded through the BMBF grant “Supercomputing and Modeling for the Human Brain” (in collaboration with the Julich Supercomputing Center (JSC)) recently joined the team of developers at IAS-5/INM-9 and is currently working towards the implementation of the path-MD algorithm within the MiMiC QM/MM interface while targeting the large-scale GPU clusters hosted at JSC. As a first step towards this goal, we have implemnted the algorithm in the GROMACS sofwtare for classical biomolecular simulations and benchmarked its performance on the JUWELS Booster (see Figure 1). This work is done in collaboration with Prof. Thomas Lippert and Prof. Estela Suarez at JSC.
As part of our research activity, we are also devoting efforts towards designing efficient statistical-mechanics-based methods for the sampling of reactive pathways (also using Machine-Learning approaches). In a recent publication, we have developed an approach that leverages on data-driven methods combined with the metadynamics of paths approach to train efficient ML-based collective variables for free energy calculations [2]. This is done in collaboration with Prof. Michele Parrinello (IIT, Genova, Italy) and Dr. Andrea Rizzi, within the Helmholtz European Partnering program.
These activities are supported also via the European Joint Doctorate program AQTIVATE.
People involved



Collaborators

Prof. Dr. Dr. Thomas Lippert
Director of Jülich Supercomputing Centre Speaker of Helmholtz Information Program 1 PI in Topics 1 and 2 & Joint Lab SMHB
- Jülich Supercomputing Centre (JSC)
Room R 360

Prof. Dr. Estela Suarez
Joint Lead of JSC-Division "Novel System Architecture Design" Spokesperson of Helmholtz Information Program 1, Topic 2, and PI in Topics 1 and 2
- Jülich Supercomputing Centre (JSC)
Room 222

Fundings

References
- Mandelli D, Hirschberg B, Parrinello M. Metadynamics of Paths (2020) Phys. Rev. Lett. 125(2): 026001. doi: 10.1103/PhysRevLett.125.026001
- Müllender L, Rizzi A, Parrinello M, Carloni P, Mandelli D. Effective Data-Driven Collective Variables for Free Energy Calculations from Metadynamics of Paths, PNAS Nexus 3(4): 159. doi: 10.1093/pnasnexus/pgae159
Applications
Some applications based on the these enhanced sampling techniques are currently in progress:
Using Metadynamics to investigate the binding of ligand furosemide with mitoNEET protein
Firstly, we applied a variant of a widely used enhanced sampling method - Metadynamics, namely Localized Volume-based Metadynamics (LV-MetaD), to study the system of furosemide binding with human mitoNEET (mNT) protein. mNT is homodimeric and contains two [2Fe-2S] clusters with a novel 3Cys:1His coordination. The mNT’s [2Fe-2S] clusters are labile. The lability is controlled by environmental pH and [2Fe-2S] cluster redox states. Aberrant release of the [2Fe-2S] cluster relates to a variety of diseases, including cancer and neurodegeneration. The binding of furosemide to mNT was shown to slow down [2Fe-2S] cluster release in vitro. However, due to the lack of well-defined binding pocket on mNT, molecular docking without considering the solvent effect may not accurate and this is why we need an enhanced sampling method.
Here in LV-MetaD, we restraint the movement of furosemide inside a parabolic-solid volume including the experimental binding site and neighboring regions, and employ three CVs to deposit bias potential: ρ, the distance between the center of mass of the furosemide and the protein, τ, the parameter that defines the parabolic-solid shape of the volume, θ, defined as the azimuthal angle of its orthogonal projection on the x-y plane (Figure 2).

Figure 2 - The volume and three CVs (ρ, τ, θ) in LV-MetaD for furosemide-mNT complex system
Our results identified multiple poses of furosemide, including the one observed in the X-ray structure, at the same shallow site on the surface as in the crystal structure (Figure 3). It might attribute to the packing effect in the crystal which is absent in an aqueous solution. The binding affinity calculated from LV-MetaD agrees with the experiment result.
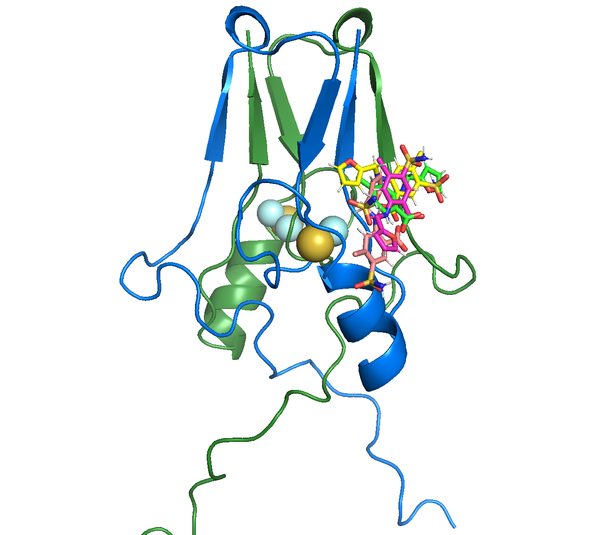
Figure 3 - Multiple poses of furosemide in binding with mitoNEET protein.
The furosemide binding predictions were carried out by imposing the binding in the [2Fe–2S] clusters region. Then we address the question of whether a ligand can also bind outside the cluster binding domain. We focus on the binding of another ligand (A, 2-benzamido-4-(1,2,3,4-tetrahydronaphthalen-2-yl)-thiophene-3-carboxylate) to mNT, for which both activity and structural information is also available. The X-ray structure of the A·mNT complex shows that A interacts also with two adjacent A·mNT complexes in the crystal. The method we use to predict the free energy landscape associated with poses all over the surface of the protein is volume-based metadynamics (Figure 4).

Figure 4 - The volume and three CVs (ρ, τ, θ) in volume-based MetaD for the A·mNT complex system.
In this case, different protonation states of the iron-bound His87 and redox states were considered. Our affinity predictions on the deprotonated and oxidized mNT turned out to be consistent with experiment in the same conditions. In an aqueous solution, where such packing forces are absent, A binds not only to the [2Fe-2S] clusters binding region but also to the L1/L3 loops and L2 loops of β-cap with multiple binding poses (Figure 5). Further, we found the binding affinity of A correlates to the intermolecular contacts formed by the drug with the protein. The simulations showed that crystal packing likely stabilizes the pose of the ligand in the cluster region at the same temperature. At physiological temperature, the redox and protonation states affect the poses and affinities. The reduction on the [2Fe-2S] clusters of mNT with deprotonated His87 allow A to reach the L2 loop. The protonation of iron-bound His87 on oxidized mNT lead to binding to β1 sheet and L1 loop. The reduction has little effect on the affinity.

Figure 5 - Multiple binding site and poses of A in binding of mNT in aqueous solution.
Our protocol can be used as a general tool for predicting multiple poses and binding affinity of ligands with other human NEET proteins and play important role in drug design targeting these proteins.
Ligands binding RNA hairpin
The aberrant interaction between RNA hairpin and proteins may be associated with a variety of diseases, including neurodegenerative and viral diseases and RNAs also play an important role in the replication of virus. We plan to study ligands which may affect RNA-protein interactions in a variety of clinical settings. As a starting project, we reproduce the binding affinity and binding pose of ligands binding to the RNA hairpin HIV-1 TAR. The work is in collaboration with Prof. G. Varani (U. Washington).
References
- Multiple Poses and Thermodynamics of Ligands Targeting Protein Surfaces: The Case of Furosemide Binding to mitoNEET in Aqueous Solution. Hoang, Linh Gia, et al. (2022). Frontiers in cell and developmental biology 10, 886568.
- Predictions of the poses and affinity of a ligand over the entire surface of a NEET protein: the case of human mitoNEET. Zuo, Ke, et al. (2023). Journal of Chemical Information and Modeling 63, 643-654.