Massively parallel QM/MM
Many quantum biological applications (e.g. simulations of enzymatic reactions) usually require dealing with large systems (typically of the order of 104 - 105 atoms), making first principles quantum mechanical strategies far too costly. This motivates using a multiscale approach (see Figure 1), in which the part of the system that is of particular interest (e.g. the active site of an enzymatic reaction) is treated at quantum level (QM part), while the rest of the system is handled by a classical force field (MM part). Such a hybrid QM/MM approach allows a significant decrease of the size of the computationally expensive part, while keeping the ability to represent the processes that can only be treated by quantum chemistry (e.g. chemical reactions). Unfortunately, most of the current implementations of QM/MM codes do not scale very well, limiting tremendously the domain of applications of this otherwise very powerful approach. Our activities are aimed at developing HPC-oriented software, combining highly-scalable QM/MM code with statistical mechanics based algorithms and machine learning techniques to study the thermodynamics and the kinetics of neurobiologically relevant systems.
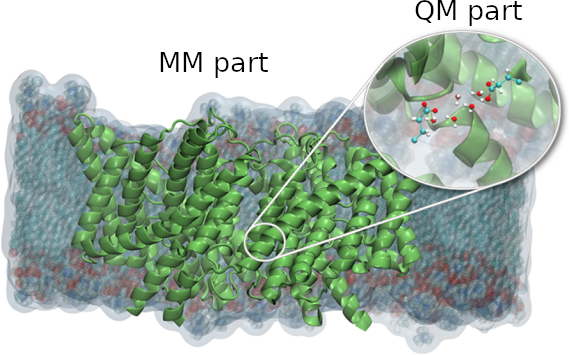
Figure 1: In multiscale atomistic QM/MM simulations, the (small) region of interest is treated at the quantum mechanical level of theory (QM part) while the rest of the system is handled by classical force field (MM part). This allows studying large biological systems (~100,000 atoms) while keeping the ability to describe processes that can only be treated by quantum chemistry.
MiMiC: A Novel Framework for Multiscale Modeling in Computational Chemistry

In an effort at coding a truly HPC QM/MM code, we are focusing on building a QM/MM coupling scheme based on the CPMD code. This is a highly efficient massively parallel first-principles (quantum) molecular dynamics software package. It can scale up to a few million threads with extremely high efficiency. The old built-in QM/MM implementation of CPMD has a set of issues that prevent its usage on large-scale neurobiological applications. First, the scalability of the MM description is limited, thus, simulating large systems becomes time consuming. Second, the number of classical force fields that can be used is restricted to AMBER and GROMOS96 formats. Finally, because of the tight coupling of CPMD to routines from the GROMOS96 code, which is used to handle the classical part of the simulation, the user needs to buy a commercial GROMOS96 license in order to be able to run QM/MM simulations.
We have recently developed a new QM/MM platform (MiMiC) [1,2], which uses a loose coupling scheme to connect CPMD with in principle any client MM code, and in particular we have explicitly coupled it to the GROMACS code [1]. The loose coupling requires the use of a communication layer in order to establish data interaction between the independent codes (see Figure 2). This approach allows us to benefit from the highly efficient parallelization schemes of both CPMD and GROMACS codes [2] (see Figure 3). It also provides us with a flexible and easily extendable framework that potentially will allow the support of any kind of MM code and any type of force field. Finally, the loose coupling allows overriding licensing issues that can arise when coupled codes have not permissive licenses.
The code has been recently applied to study the molecular basis of CLC antiporter inhibition by fluoride [3] and the mechanisms of proton release [4] by performing sub-nanosecond DFT-based QM/MM simulations, to investigate the accuracy of molecular simulation-based predictions of koff values of a ligand iperoxo targeting the muscarinic receptor M2 [5], and to study enzymatic reactions [6]
This MiMiC project is performed in collaboration with the group of Prof. U. Rothlisberger (EPFL Lausanne, Switzerland), Prof. J. M. Haugaard Olsen (University of Southern Denmark, Odense M, Denmark) and Prof. S. Meloni (University of Ferrara, Italy), and it has been supported by the European BioExcel Center of Excellence and the European Joint Doctorate HPC-LEAP.
Current efforts are dedicated to further improving the scalability of the code, also using machine learning and via the implementation of statistical-mechanics based free energy methods.
These activities will be supported also via the European Joint Doctorate program AQTIVATE (Applications are opened for 3 PhD positions! More info here).
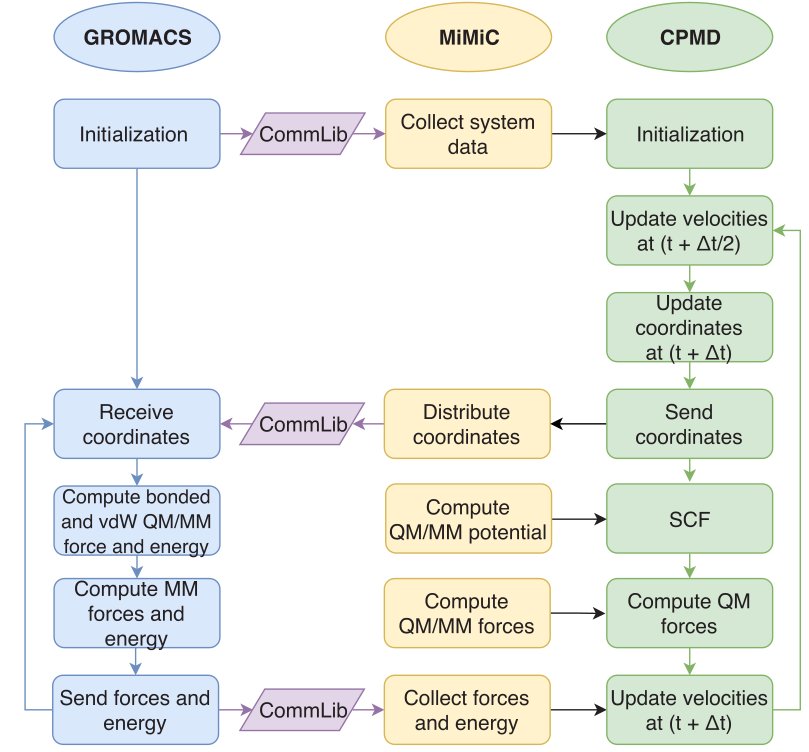
Figure 2 - Illustration of the MiMiC-based QM/MM Born-Oppenheimer MD workflow using the velocity Verlet algorithm.

Figure 3 - Using CPMD and GROMACS as QM and MM engines - coupled via MiMiC - we demonstrated scaling up to more than 80,000 CPU cores with efficiency about 70% using hybrid functionals to perform QM/MM simulations of very large biological systems [6].
People involved

Prof. Dr. Paolo Carloni
Director

Dr Davide Mandelli
Researcher

Dr. Emiliano Ippoliti
Staff member and IT coordinator
References
- Olsen J M H, Bolnykh V, Meloni S, Ippoliti E, Bircher M P, Carloni P, Rothlisberger U. (2019) MiMiC: A Novel Framework for Multiscale Modeling in Computational Chemistry. J. Chem. Theory Comput. 15(6): 3810-3823. doi: 10.1021/acs.jctc.9b00093
- Bolnykh V, Olsen J M H, Meloni S, Bircher M P, Ippoliti E, Carloni P, Rothlisberger U. (2019) Extreme Scalability of DFT-Based QM/MM MD Simulations Using MiMiC. J. Chem. Theory Comput. 15(10): 5601-5613. doi: 10.1021/acs.jctc.9b00424
- Chiariello M G, Bolnykh V, Ippoliti E, Meloni S, Olsen J M H, Beck T, Rothlisberger U, Fahlke C, Carloni P. (2020) Molecular Basis of CLC Antiporter Inhibition by Fluoride J. Am. Chem. Soc. 142(16): 7254-7258. doi: 10.1021/jacs.9b13588
- Chiariello M G, Alfonso-Prieto M, Ippoliti E, Fahlke C, Carloni P. (2021) Mechanisms Underlying Proton Release in CLC-type F-/H+ Antiporters J. Phys. Chem. Lett. 12(18): 4415-4420. doi: 10.1021/acs.jpclett.1c00361
- Capelli R, Lyu W, Bolnykh V, Meloni S, Olsen J M H, Rothlisberger U, Parrinello M, Carloni P. (2020) Accuracy of Molecular Simulation-Based Predictions of k off Values: A Metadynamics Study J. Phys. Chem. Lett. 11(15): 6373-6381. doi: 10.1021/acs.jpclett.0c00999
- Raghavan B., Paulikat M., Ahmad K., Callea L., Rizzi A., Ippoliti E., Mandelli D., Bonati L., De Vivo M., and Carloni P. (2023) Drug Design in the Exascale Era: A Perspective from Massively Parallel QM/MM Simulations. J. Chem. Info. Mod. 63(12): 3647-3658. doi: 10.1021/acs.jcim.3c00557
Applications
Several applications based on MiMiC code are currently in progress:
Investigation of the IDH1 catalytic mechanism
The highly scalable MiMiC QM/MM package, recently developed at FZJ in collaboration with other European institutes, will be used to provide insights on the catalytic mechanism of the Isocitrate Dehydrogenase 1 enzyme (IDH1). This enzyme is involved in grade 2 and 3 glioma and also some cases of glioblastoma. The study will be one of the first applications of a highly scalable HPC based approach to the investigation of such a large enzymatic system at the QM/MM level. The understanding of the mechanism of catalysis might facilitate the development of transition state analogues that would function as inhibitors of the cancer-causing IDH1 enzyme.
- Katya
- Florian
- Mirko
- Samira