Hybrid molecular mechanics/coarse grained approach
Ligand-protein docking is currently an important tool in drug discovery efforts; indeed, in the last years, structure-based drug design protocols have been the subject of important developments in general, and in particular in neurobiology. Despite the high levels of accuracy of computational tools, there are still important challenges if we want to deeply characterize the mechanism of interaction between neuronal receptors and their cognate ligands. Undertaking these challenges is of fundamental importance on protein classes of the upmost neurobiological relevance like G-protein coupled receptors (GPCRs), which constitute as much as 30% of the overall proteins targeted by FDA-approved drugs. Unfortunately, the lack of experimental structural information for most human GPCRs (hGPCR, more than 90%) along with low sequence identity (SI) across these proteins (often below 30%) have hampered rational design efforts for many highly promising hGPCR drug targets. This condition strongly limits the predictive power of computer-aided structural approaches because traditional homology modeling techniques applied to proteins with low SI with their template may lead to rather inaccurate models. All-atoms molecular dynamics (MD) simulations, very successful in refining high-quality homology-based models, may fail to improve the predictions and even lead to partial unfolding, if one starts from structures with highly inaccurate side-chain orientations as encountered in very low-resolution protein models. An alternative strategy to address this issue consists of including the minimum number of degrees of freedom of the system for the specific problem that one has in mind, leaving out unreliable information that could bias the simulation results. Keeping this strategy, our Institute have adapted a hybrid molecular mechanics/coarse-grained (MM/CG) approach, previously developed for enzymes [1], to GPCR/ligand complexes [2,3].

Within this scheme, only the region of interest (the binding site along with the ligand and the solvation water) is treated at molecular mechanics (MM) level using an atomistic force field, while the rest of the system is described at coarse grained (CG) level.
In the CG region, each residue is modeled through a bead centered on the Cα and interacting with the other beads through a Gō-type potential). Specifically, the bonded interactions are represented by a quartic potential, while the non-bonded interactions are represented by a Morse potential. An intermediate region (I) between the MM and the CG domains ensures the coupling between the two levels of description and the protein backbone integrity. "I" residues are described at atomistic level, and their Cα and Cβ atoms interact with CG beads according to the CG potential. The selection of the atoms in the MM, I, or CG regions does not require updates during the simulation.
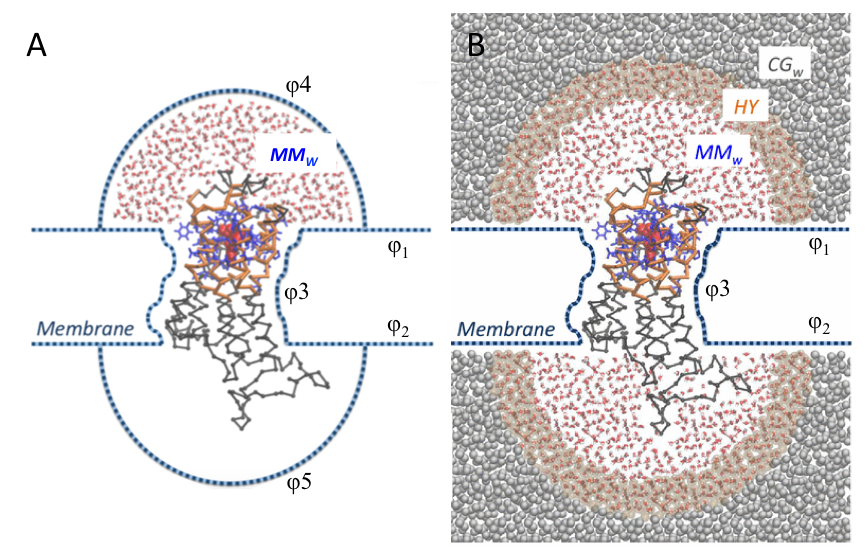
The presence of the membrane is implicitly modeled as a series of boundary potentials: two repulsive potentials placed at the level of the lipids’ heads prevent penetration of water molecules in the membrane space (𝛗1 , 𝛗2) while a softened Lennard-Jones-like potential (2-1) acting on transmembrane residues’ Cα (𝛗3) mimics the interactions between the protein and the membrane. As for the solvent, in the initial version of MM/CG, water evaporation from the atomistic region was prevented through a repulsive potential, which confines water molecules in a hemispherical region around the binding site (𝛗4 , 𝛗5) (Figure2A).
We have developed the Hybrid MM/CG webserver [4] to set up and run short MM/CG simulations.
In order to improve solvent description and remove potential artifacts originating from the confinement of the solvation water, we have recently implemented a multiscale representation of the solvent, based on an adaptive resolution scheme in the Hamiltonian formulation (H-AdResS) [4,5] (Figure 2B). This allows water molecules to freely diffuse between fully atomistic (MMw) and coarse-grained (CGw) domains, maintaining a uniform density between the two while changing on-the-fly their resolution. The MMw regions, where atomistic water is described by the SPC/E potential (VwMM), are shaped as hemispheres capping the extracellular and intracellular domains of the protein, whereas coarse-grained water is present outside of these boundaries in a bigger box (CGw). Coarse-grained water is modeled by beads coinciding with the center of mass of each molecule, interacting through a potential, VwCG, derived from fully atomistic simulation of pure water at the same state point through iterative Boltzmann inversion. The smooth coupling between the two domains is achieved by introducing an intermediate hybrid region (HY), where the potentials describing the MMw and CGw models are linearly combined through a switching function λα = λ(Rα), which depends on the instantaneous position Rα of the center of mass of the α water molecule. Specifically, λα smoothly changes from 1 to 0 when moving from MMw to CGw boundaries. A correction term is added to water molecules in the HY region to ensure uniform density across the MMw, HY and CGw domains. This term preserves a constant chemical potential, leading to the simulation of a grand canonical ensemble in the high-resolution region. In this respect, the CGw region can be regarded as a reservoir of coarse-grained water molecules, and we call the overall setup open-boundary (OB)-MM/CG.
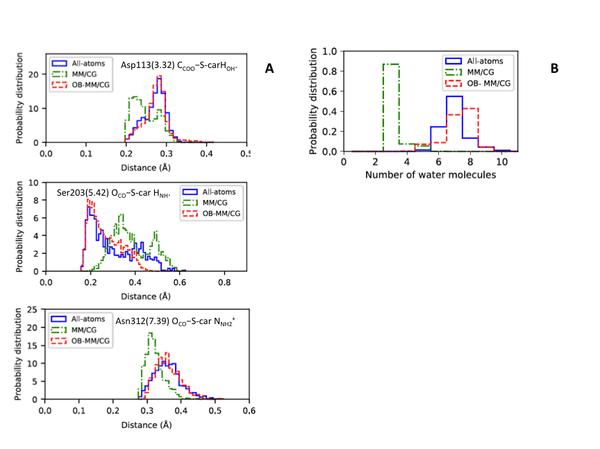
The new OB-MM/CG scheme has been validated on a well-studied GPCR, the β2-adrenergic receptor, in complex with its inverse agonist S-carazolol. Structural and dynamical properties of both the solvent and the complex in the OB-MM/CG simulations are in good agreement with results from fully atomistic simulations. Although the original MM/CG scheme reproduces ligand poses fairly well, the more realistic hydration achieved through OB-MM/ CG leads to sizable improvements in the description of the binding configurations explored by the ligand/protein complex (Figure 3) and of the binding site flexibility. These results provide solid ground for the use of the OB-MM/CG scheme for binding free energy calculations in GPCR–ligand complexes (work in progress).
People involved

Prof. Dr. Paolo Carloni
Director

Prof. Alejandro Giorgetti
Associated member

Dr. Mercedes Alfonso-Prieto
Group leader

Dr. Vania Calandrini
Group leader

Dr. Emiliano Ippoliti
Staff member and IT coordinator
References
- Neri M, Baaden M, Carnevale V, Anselmi C, Maritan A, et al. (2008) Microseconds Dynamics Simulations of the Outer-Membrane Protease T. Biophysical Journal. 94: 71–78. doi: 10.1529/biophysj.107.116301
Leguebe M, Nguyen C, Capece L, Hoang Z, Giorgetti A, et al. (2012) Hybrid molecular mechanics/coarse-grained simulations for structural prediction of G-protein coupled receptor/ligand complexes. PLoS ONE 7: e47332. doi: 10.1371/journal.pone.0047332
Schneider J, Korshunova K, Si Chaib Z, Giorgetti A, Alfonso-Prieto M, Carloni P (2020) Ligand pose predictions for human G protein-coupled receptors: insights from the Amber-based hybrid Molecular Mechanics/Coarse-Grained approach. J. Chem. Inf. Model., 60(10): p. 5103-5116. doi: 10.1021/acs.jcim.0c00661
- Schneider J, Ribeiro R, Alfonso-Prieto M, Carloni P, Giorgetti A (2020) Hybrid MM/CG Webserver: Automatic set up of molecular mechanics/coarse-grained simulations for human G protein-coupled receptor/ligand complexes.Front. Mol. Biosci., 7: p. 576689. doi: 10.3389/fmolb.2020.576689
Tarenzi T, Calandrini V, Potestio R, Giorgetti A, Carloni P (2017) Open boundary simulations of proteins and their hydration shells by Hamiltonian adaptive resolution scheme. Journal of chemical theory and computation 13(11): p. 5647-5657. doi: 10.1021/acs.jctc.7b00508
Tarenzi T, Calandrini V, Potestio R, Carloni P (2019) Open-Boundary Molecular Mechanics/Coarse-Grained Framework for Simulations of Low-Resolution G-Protein-Coupled Receptor–Ligand Complexes. Journal of chemical theory and computation 15(3): p. 2101-2109. doi: 10.1021/acs.jctc.9b00040
Applications
Some applications based on the MM/CG approach are currently in progress:
- Nicolas