Machine-learning-accelerated molecular simulations
Many biologically- and pharmaceutical-relevant molecular events such as enzymatic reactions and drug-protein dissociations can take place on timescales reaching seconds to hours, which are out of reach for molecular dynamics (MD) simulations using hybrid quantum mechanics/molecular mechanics (QM/MM) potentials. Machine learning techniques provide an effective paradigm to analyze QM/MM data and extract information about the relevant physics that we can in turn exploit to study slow molecular processes.
Accurate free energies with normalizing flows
Predicting the free energy surface (FES) is a task of fundamental importance for the characterization of equilibrium and dynamical properties of a molecular process (e.g., binding or enzymatic/chemical reactions). We are developing schemes that enable the calculation of the FES at highly accurate (and expensive) levels of quantum mechanical theory. This is made possible by a combination of enhanced sampling simulations (e.g., metadynamics) and normalizing flow neural networks, which are trained to accelerate the calculation of free energies from data obtained from cheaper (but less accurate) potentials [1]. The method is highly scalable and exploits the parallelization capabilities of our QM(/MM) software.
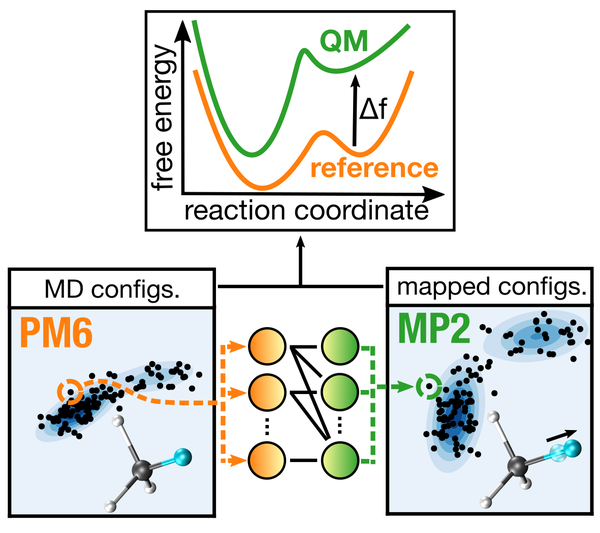
Figure 1 - Schematics of the targeted free energy perturbation methodology [1] used to obtain a free energy profile at the MP2 quantum mechanical level of theory from simulations using the semi-empirical PM6 potential.
Machine learning collective variables for enhanced sampling
The dynamics of molecular events is often characterized by large energetic barriers separating two (or more) states of interest (e.g., reactants and products, bound and unbound protein-ligand complexes). The time required to observe such transitions increases exponentially with the height of the barrier. In many molecular processes in the cell, barriers are too high to simulate spontaneous transitions using plain molecular dynamics simulations. Enhanced sampling techniques such as metadynamics are capable of accelerating specific events by reducing the height of the barrier in the direction of a low-dimensional collective variable (CV). Such CVs can be, however, extremely challenging to determine and typically require extensive knowledge of the system. In collaboration with the group of Prof. Michele Parrinello at the Italian Institute of Technology (IIT), we are developing data-driven methodologies that can result in very effective collective variables that are capable of well describing the slow modes of complex molecular processes such as protein-drug unbinding.
Neural network interatomic potentials for biological systems
Molecular simulations are often subject to a trade-off between the size/timescale of the molecular event and the accuracy of the potential energy function model describing the physics of the system. On one end, empirical potentials (e.g., force fields) are sufficiently cheap to enable the study of large macromolecules such as proteins. These models, however, are not always transferable, for example to systems where electronic polarization plays an important role. On the other, quantum mechanical potentials provide a more general description of the system’s physics but can be prohibitively expensive. In recent years, machine learning models of the potential have emerged as a third viable paradigm. We are developing and exploiting neural network models of the potential energy function that enable near-DFT accuracy at a fraction of the computational cost while maintaining scalability to large heterogeneous systems such as small molecules and proteins in solution.
These activities will be supported also via the European Joint Doctorate program AQTIVATE (Applications are opened for 3 PhD positions! More info here).
People involved



Collaborators


Dr. Katya Ahmad
Science Manager at TUM Chair of Proteomics and Bioanalytics (GERMANY)
Fundings

References
- Rizzi A, Carloni P, Parrinello M. (2021) Targeted free energy perturbation revisited: Accurate free energies from mapped reference potentials. J. Phys. Chem. Lett. 12(39): 9449-9454. doi: 10.1021/acs.jpclett.1c02135