Origins, rise to prominence and future of Density Functional Theory reviewed
05 October 2015
In little more than 20 years, the number of applications of density functional (DF) formalism in chemistry and materials science has grown in an astonishing fashion. The number of publications alone shows that DF calculations have proved hugely successful, which is even more remarkable considering the fact that the widespread application of density functional methods, particularly in chemistry, only began after 1990. The origins of DF calculations are, however, usually traced to the papers of Hohenberg, Kohn, and Sham more than a quarter of a century prior to this rapid gain in acceptance of these methods. In the late eighties, the combination of DF calculations with molecular dynamics promised to provide an efficient way to study structures and reactions in molecules and extended systems.
Dr. Robert O. Jones from Quantum Theory of Materials (PGI-1) has recently reviewed the development of density-related methods back to the early years of quantum mechanics and follows the breakthrough in their application after 1990. In the prestigious journal Reviews of Modern Physics, he presents two examples from biochemistry and materials science, which are among the many current applications that simply exceeded all expectations in 1990. Furthermore, he discusses the reasons why some of the most cited practitioners in the field are currently concerned about its future.
Original publication:
Density functional theory: Its origins, rise to prominence, and future
R. O. Jones,
Reviews of Modern Physics
, Vol. 87, July – September 2015, published 25 August 2015DOI: 10.1103/RevModPhys.87.897
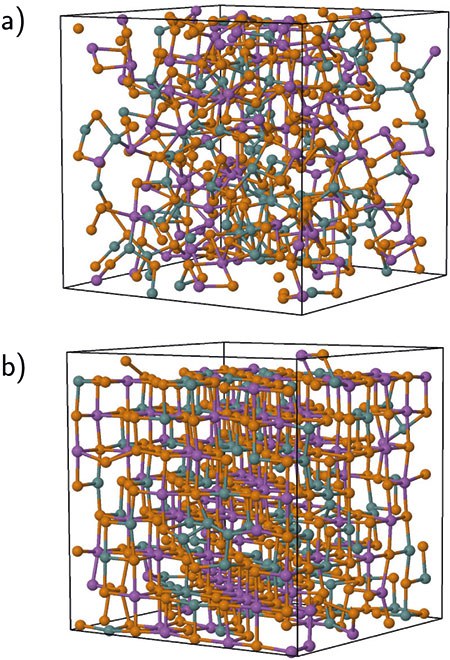
Example of a DFT application: Simulation of the crystallization in Ge2Sb2Te5 (GST) alloy at 600 K. Such alloys are often used in information technology. (a) Amorphous structure after 215 ps, (b) crystalline structure. Crystallization takes place in just over 1 ns, and it is possible to monitor changes in the spatial distribution of the cavities during this time. The presence of cavities is essential for rapid crystallization in these materials. These calculations required over 400,000 self-consistent DF calculations of energies and forces for a 460-atom sample, after 1045 ps.