Materials Lab Computer
Phase Change Materials: the Future of Computer Memory?
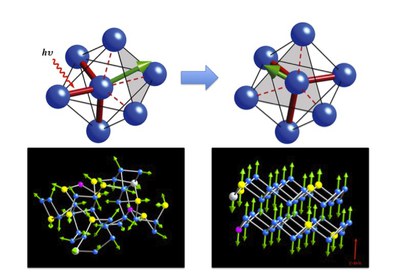
Phase change (PC) materials remain leading candidates for future computer random access memory (RAM) and rewritable storage devices (CD-RW, DVD-RW, Blu-ray Discs, etc.). The battle to replace the digital versatile disk (DVD) was decided in 2008 in favour of the Blu-ray Disc, and the recording media of all BD products involve PC materials. Information is stored in these devices in the form of microscopic bits (each less than 100 nanometers in size) in an ultrathin layer of a polycrystalline alloy containing several elements. The bits may have a disordered (amorphous) or an ordered (crystalline) structure, and the transition between the two phases is not only extremely rapid (some tens of nanoseconds) but also reversible. Amorphous bits are formed by quenching after a localized and short (~ 1 nanosecond) pulse to heat them to a temperature above their own melting point. Longer laser heating (of the order of 10 ns) to above the glass transition but below the melting point returns the bit to the metastable crystalline form. The state can be identified by monitoring the optical or electrical properties..
The physical requirements of PC materials, particularly the rapid crystallization, are satisfied by relatively few materials. The focus for some years now has been on alloys of three or four elements, many of which contain germanium (Ge), antimony (Sb) and tellurium (Te) ["GST alloys", common in BD applications] or alloys of Sb (70%) and Te (30%). With small amounts of silver (Ag) and indium (In), "AIST" alloys are in widespread use in DVD-RW devices.
Although both alloy families contain Sb and Te, the phase change mechanisms are very different. In GST materials, the amorphous bit crystallizes via nucleation, i.e. small crystallites formed in the interior grow rapidly until they cover the whole bit. The phase change in AIST alloys proceeds from the rim of the bit adjoining the crystalline surroundings, towards its interior.
We have performed simulations of the amorphous structure of prototype members of both families and have presented plausible scenarios for the crystallization of each. In AIST, we propose a "bond exchange" model in which the local environment in the amorphous bit is changed by small movements of an Sb atom that result in the exchange of a "short" and a "long" bond. A sequence or avalanche of many such steps results in reorientation (crystallization) without requiring large atomic motions.
R. O. Jones, J. Akola
Biologische und organische Reaktionen
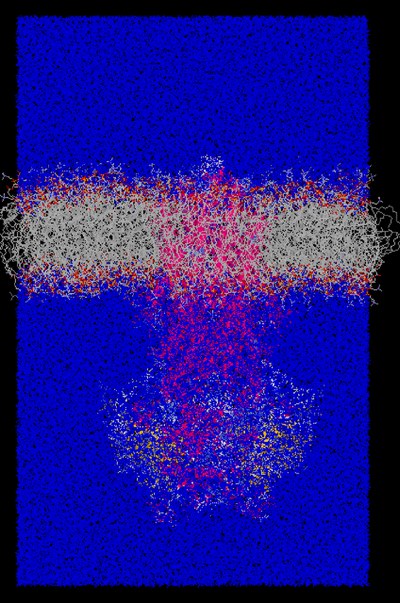
Die Kombination von Biologie und Informatik gehört zu den größten Wachstumgebieten der Wissenschaft, und die Verfügbarkeit von massiv parallelen Rechnern hat einen wichtigen Beitrag geleistet. Trotz der vielen Anwendungen bei der Entwicklung von Arzneien, der Bestimmung der Gene in DNS-Sequenzen und der Proteinstrukturen von deren Sequenzen, bleibt jedoch für die Untersuchung von Reaktionen in biologischen Molekülen auf *atomistischen* Längenskalen noch viel zu tun. Der Dichtefunktionalformalismus liefert prinzipiell ein Mittel, um solche Probleme zu untersuchen, aber selbst die leistungsfähigsten Rechner von heute können kaum mehr als 1000 Atome auf Zeitskalen von einigen Hundert Pikosekunden behandeln. Sie taugen kaum für die meisten Probleme in der Biologie, wo Zehntausende Atome über Mikrosekunden oder viel länger behandelt werden müssen. Solche Probleme verlangen die Anwendung von klassischen Kraftfeldern, und wir haben sie in einer Untersuchung von ABC-Transportern (Adenosin 5'-Triphosphat Bindungs-Kassetten-Transporter) benutzt. ABC-Transporter sind Membranproteine, die aktiv Substrate durch Lipid-Bischichten transportieren, und das Protein Sav1866 gehört dazu. Sav1866 ist durch die Analyse von einer Form des Bakteriums staphylococcus aureus entdeckt worden, die gegen Antibiotika resistent war. Wir haben die Struktur von Sav1866 in einer Lipid-Bischicht mittels Simulationen von ca. 200000 Atome über Zeiten von über 1 Mikrosekunde untersucht.
R. O. Jones, J. Akola, J.-H. Lin (Taipei)
Organic Molecules on Surfaces - Molecular Electronics
The idea of exploring and monitoring new possibilities of incorporating organic molecules into existing technologies and building molecule-based nanoscale electronic circuits with rectifying, logic, and switching functions has stimulated experimental and theoretical efforts to study and predict their properties. The development of organic/inorganic interfaces depends critically on understanding the bonding and lateral interactions that govern the orientation, conformation, and two-dimensional arrangement of molecules at surfaces.
Density functional (DFT) calculations provide new insight into this problem, particularly the interactions between such molecules, and we have used the EstCoMPP program, a projector augmented plane-wave package, to investigate the geometrical and electronic structure of several molecular layers that use the carboxylate group as an anchor to metal surfaces.
Organic Molecules on Surfaces - Molecular Electronics

The DF method is capable of determining energies as a function of atomic positions, and it should be able in principle to predict mechanical and thermo-mechanical properties, such as thermal expansion coefficients or elastic constants of crystalline materials. The theory of thermal expansion developed by Born and Grüneisen requires knowledge of the phonon frequencies and the way they change with changing unit cell size. This is a great computational challenge and our success in explaining the thermal expansion of ß-eucryptite - the basic material from which all cooktops are made - is quite remarkable.