Simulations of Top7
In the section "Folding of Top7-CFr" of this gallery, we show our simulations of the C-terminal fragment of the protein Top7 protein. Following our study of the C-terminal fragment, we also performed MC simulations of the Top7 molecule (PDB id: 1QYS). Top7 is a designed protein and is extremely stable. It's native structure consists of two alpha helices and a 5 stranded beta sheet, in a β–β–α–β–α–β–β fold that does not occur in nature. Top7 is experimentally known to be extremely stable for its size, with a folding transition that deviates from a typical two state behaviour of common for natural single domain proteins. It also has a very slow folding rate indicating folding time of the order of 1 second. This slow folding protein is very challenging to study using computer simulations, especially with molecular dynamics simulations.
Our simulations of Top7, like all our folding simulations, started from random initial conformation of the molecule, i.e., with random values assigned to each torsional degree of freedom in our model. The parallel tempering simulations found the native folded state of Top7 regularly, and we included 25 independent (different replica in the parallel tempering simulations) folding events in the publication reporting our study.
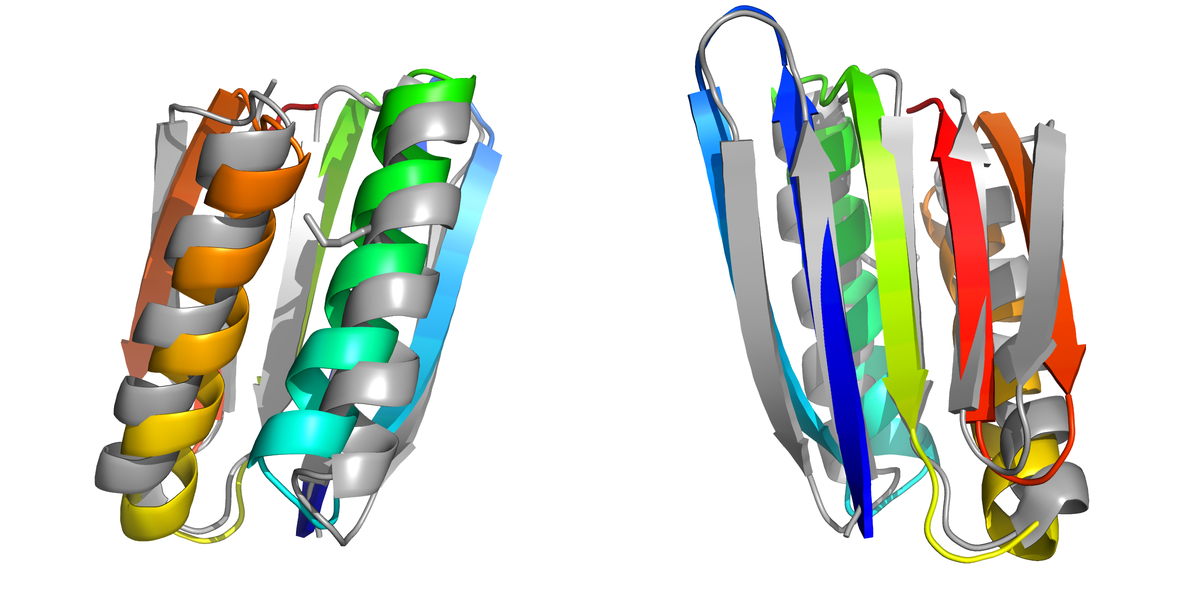
Above: Two views of the lowest energy structure observed in the simulations aligned with the experimentally determined native structure (PDB id : 1QYS). The simulation structure is shown in colours where as the PDB structure is shown in grey.
The molecule proved to be extremely stable in our simulations as well, since, once folded, it was only rarely observed to unfold. The native state was the state with the lowest energy among a very large number of low lying deep local minima. This made the simulations sluggish. Although portions of the molecule would fold into their native-like secondary structures, they would randomly attach in different orders, producing these low lying local minima.


The ensemble sampled at the lowest temperature in the simulations consisted of many compact, secondary structure rich, low energy structures. Above, we see the free energy landscape parametrised by the backbone RMSD and the radius of gyration (left) and the structural similarity measure Q (right). The native structure had the lowest energy, but yet, because of the very low lying non-native minima, diffusion to the native state from our random initial conformations was extremely slow.
Since, the proper tertiary structure arrangement, when found, was energetically more favourable, so that over time, the population of the native state increased in our simulation. Because of this, our simulations can not be said to have reached thermodynamic equilibrium for Top7, making it difficult to estimate temperature dependent properties. It is interesting, however, to note the similarity of the slow diffusion to the native state in the simulations to the rather slow folding of this protein, and the extreme observed stability in both the experiments and the simulations.
References
- "Folding of Top7 in unbiased all-atom MonteCarlo simulations", Sandipan Mohanty, Jan H. Meinke, and Olav Zimmermann, Proteins 2013; 81:1446–1456