Green's functions methods in Materials Science
One of the most important topics in today’s societal discourse is the production and consumption of energy, with the quest for microelectronics and photovoltaic devices with higher energy (conversion) efficiency as one of the core elements of a technology-based solution. As technological advances in the design and production of these devices are largely based on novel nanotechnological approaches, they highly benefit from numerical simulations at the nanoscale level. There, the most advanced device simulation methods in terms of physical validity and predictive power are based on the so- called non-equilibrium Green’s function formalism (NEGF), which, however, comes at a very large computational cost.
In order to effectively model and simulate nanoscale devices, the mathematical model and its algorithmic counterpart have to account for an out-of-the-equilibrium quantum description of an open physical system. Inelastic scattering dominates the quantum mechanical interaction between charge carriers with phonons (lattice vibrations) and photons over a large range of energies. The resulting quantum transport is non-ballistic and the formalism is developed for non-equilibrium steady state operation, which is the main mode of operation for photovoltaic devices Charge carriers flowing in and out of the device, temperature gradients, applied bias and energy balance are all elements that enforce open boundary conditions in the electronic sense, i.e., only for the electronic degree of freedom, open boundaries are implemented. In addition, the atomistic simulation of devices requires supercells with a very large number of atoms, inclusion of realistic defects and non uniform nanoscale structures.
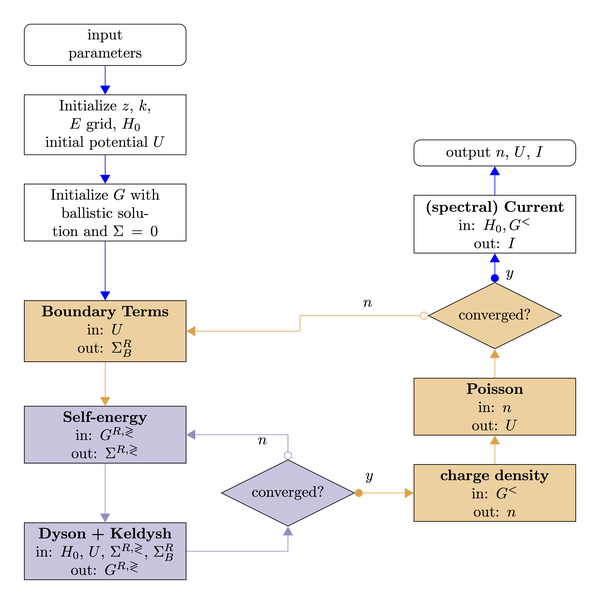
Physical systems with such characteristics can be mathematically modeled using the non-equilibrium Green’s functions (NEGF) formalism, leading to an interdependent array of equations that have to be solved self-consistently. The self-consistency is not an inherent requirement in NEGF, but originates from the way by which interactions are treated in the present approach, i.e., self-consistent Born approximation (SCBA) for the self-energies and coupling to Poisson equation. The resulting computational structure consists of two nested self-consistent iteration loops which necessitate large amounts of computing time and volatile memory even for relatively small sized simplistic systems. Thus, the simulation of realistic devices calls for the unavoidable use of large computing platforms which, in turn, demands for the parallelization of NEGF-based algorithms, which poses several challenges.
Mathematically, the full NEGF picture consists of a set of coupled integro-differential equations for the non- equilibrium Green’s functions and self-energies on a complex time contour. In the context of photovoltaic applications, where the relevant regime of operation is the steady state, the time dependence of the NEGF is simplified and transformed into an energy dependence. Performing spatial discretization by using a localized basis set, the mathematical problem is reduced to the solution of linear systems for the retarded Green’s functions (Dyson’s equation) and the correlation functions (Keldysh equations). Solving these equations together with the expressions for the interaction and contact self-energies leads to an inner self-consistent loop; requiring consistency with the solution of the Poisson’s equation for the electrostatic potential introduces an additional outer self-consistent cycle.
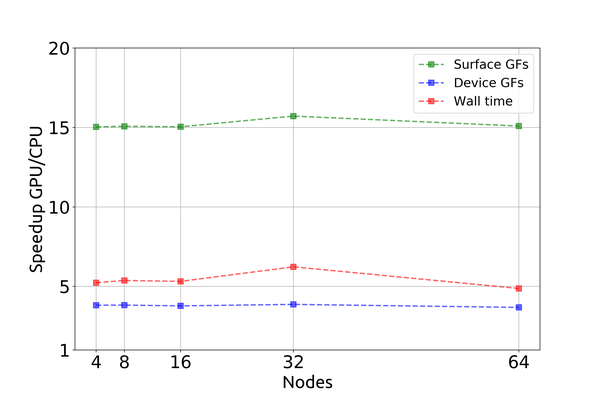
Despite the diversity of the obstacles, our past and current work shows that when correctly tackled, such composite challenge can be addressed and overcome, paving the way to state-of- the-art software (e.g., libNEGF) capable of successfully exploiting the largest supercomputing platforms currently available to simulate nanoelectronic devices with unprecedented accuracy and predictive power.