Tomographic identification of all molecular orbitals in a wide binding-energy range
Authors: A. Haags, D. Brandstetter, X. Yang, L. Egger, H. Kirschner, A. Gottwald, M. Richter, G. Koller, F. C. Bocquet, C. Wagner, M. G. Ramsey, S. Soubatch, P. Puschnig and F. S. Tautz
Phys. Rev. B 111, 165402 – Published 2. April 2025
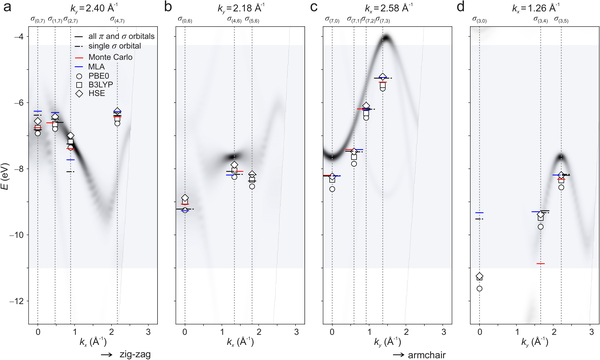
Abstract: In the past decade, photoemission orbital tomography (POT) has evolved into a powerful tool to investigate the electronic structure of organic molecules adsorbed on surfaces. Here we show that POT allows for the comprehensive experimental identification of all molecular orbitals in a substantial binding energy range of more than 10 eV. Making use of the angular distribution of photoelectrons as a function of binding-energy, we exemplify this by extracting an orbital-resolved projected density of states for 15 𝜋 and 23 𝜎 orbitals from the experimental data of the prototypical organic molecule bisanthene (C28H14) on a Cu(110) surface. These experimental results for an essentially complete set of orbitals within the given binding-energy range serve as stringent benchmarks for electronic structure methods, which we illustrate by performing density functional calculations employing four frequently used exchange-correlation functionals. By computing the respective molecular-orbital-projected densities of states, a one-to-one comparison with experimental data for an unprecedented number of 38 orbital energies became possible. The quantitative analysis of our data reveals that the range-separated hybrid functional HSE performs best for the investigated organic/metal interface. At a more fundamental level, the remarkable agreement between the experimental and the Kohn-Sham orbital energies over a binding-energy range larger than 10 eV suggests that—perhaps unexpectedly—Kohn-Sham orbitals approximate Dyson orbitals, which would rigorously account for the electron extraction process in photoemission spectroscopy but are notoriously difficult to compute, in a much better way than previously thought.
Contact

Dr. Anja Meier
Scientific Coordinator at Peter Grünberg Institute (PGI-3)
Last Modified: 13.02.2026