What glutamate transporters can reveal about rare epilepsies
What glutamate transporters can reveal about rare epilepsies
New insights from Jülich and Copenhagen offer therapeutic potential
2 July 2025
Why do some children with a particular genetic defect develop severe epileptic symptoms, while others with the same affected gene experience much milder forms of the condition? An international study involving the Institute of Molecular and Cellular Physiology (IBI-1) at Forschungszentrum Jülich in Germany has provided new answers – and opened the door to novel treatment strategies for severe early-onset epilepsy. The study was published in the renowned journal eBioMedicine and received the “Paper of the Quarter” award from the Research4Rare network, which is funded by the German Federal Ministry for Research, Technology and Space (BMFTR, formerly BMBF).
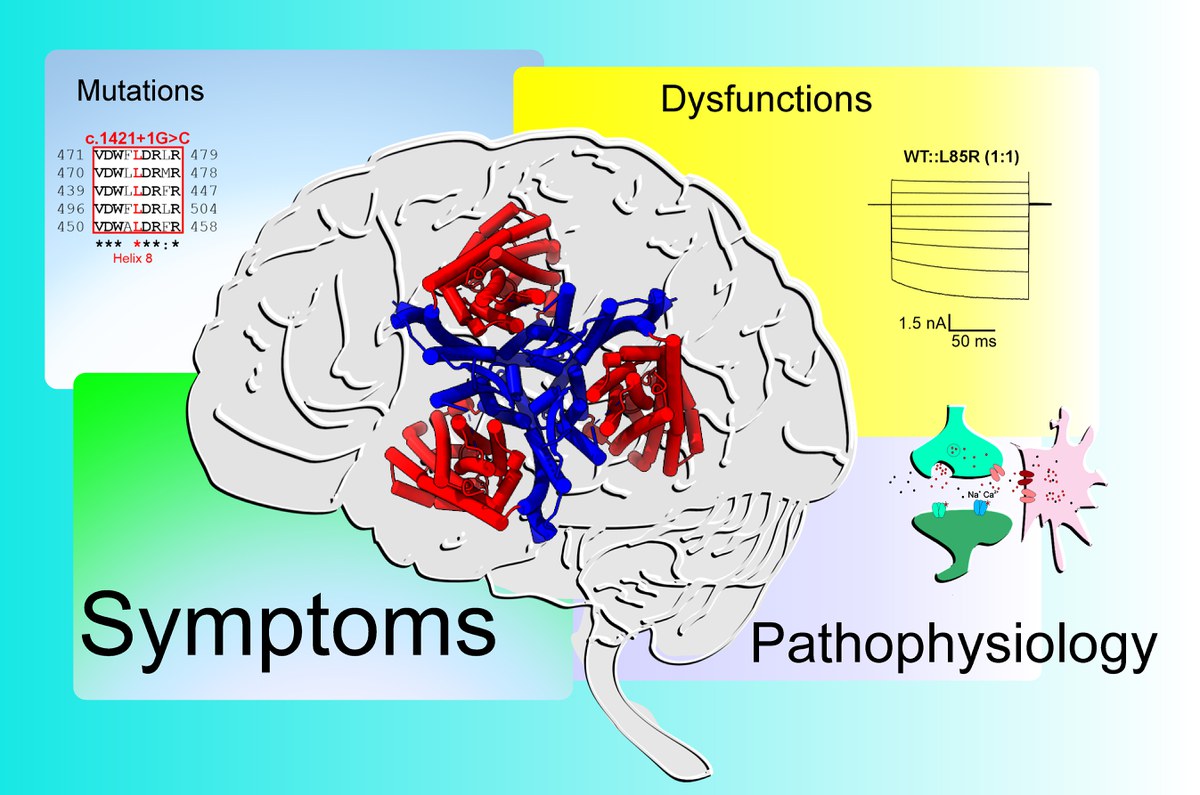
This figure illustrates the interrelationships between small subcellular changes (mutations), the functional changes they cause in the encoded protein EAAT2 (structure: in the centre of the figure), and their (patho)physiological effects with the symptoms observed in patients. | Copyrights: Forschungszentrum Jülich / Peter Kovermann
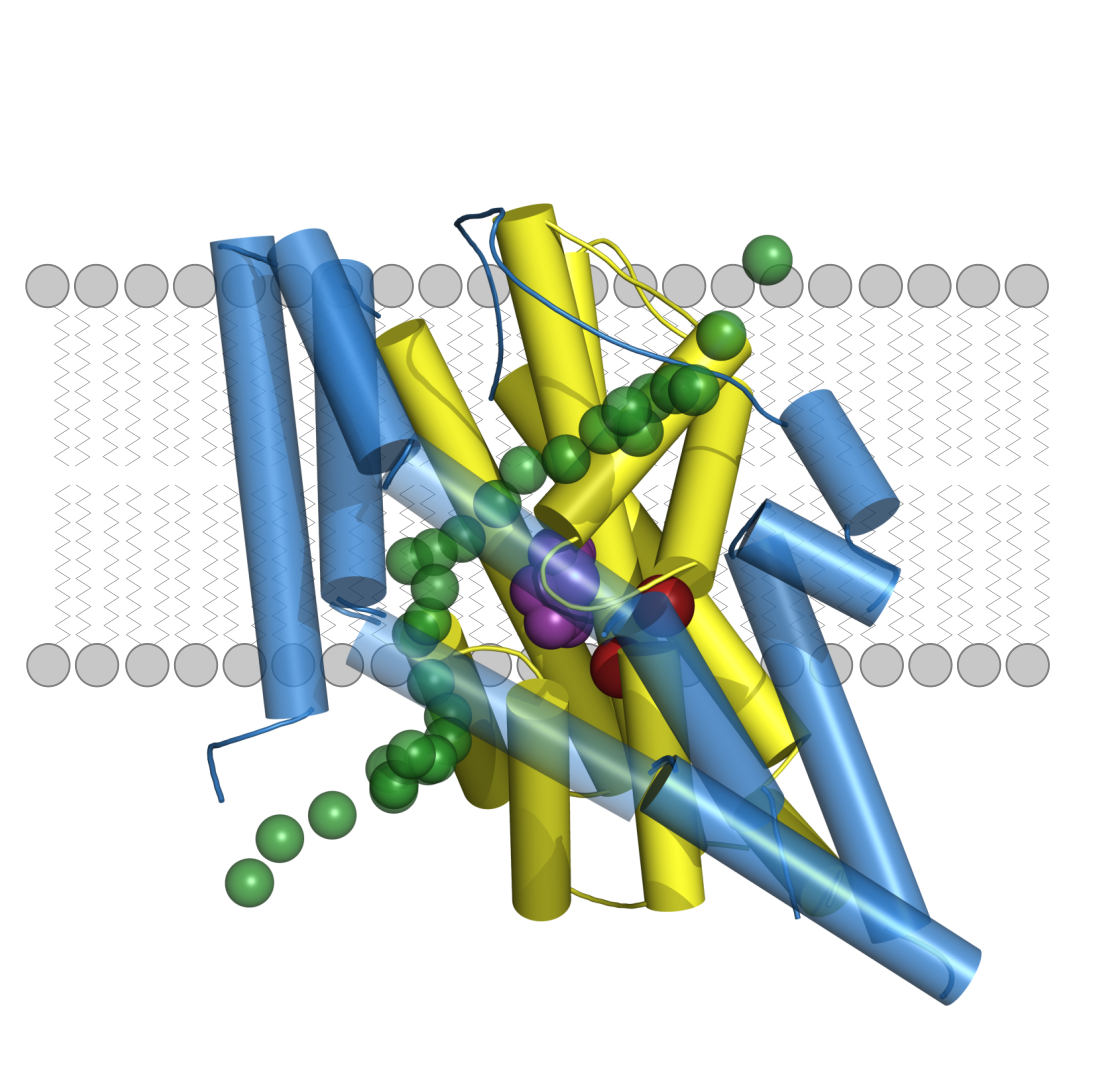
At the centre of the investigation is the SLC1A2 gene, which encodes EAAT2, the major glutamate transporter in the human brain. This transporter ensures that the neurotransmitter glutamate is efficiently removed after synaptic signalling. If glutamate is not cleared promptly from the synaptic cleft, it can lead to overstimulation of nerve cells. In addition, EAAT2 also functions as an anion channel, thereby influencing electrical signalling and ionic homeostasis in glial and neuronal cells.
Scientific focus of this study
Working with an international consortium of physicians and scientists from IBI-1 at Forschungszentrum Jülich and the Department of Drug Design and Pharmacology at the University of Copenhagen analysed 18 cases with SLC1A2 mutations to investigate their effects on EAAT2 function. They carried out functional studies in mammalian cell models. The results showed that not all mutations impaired glutamate transport.
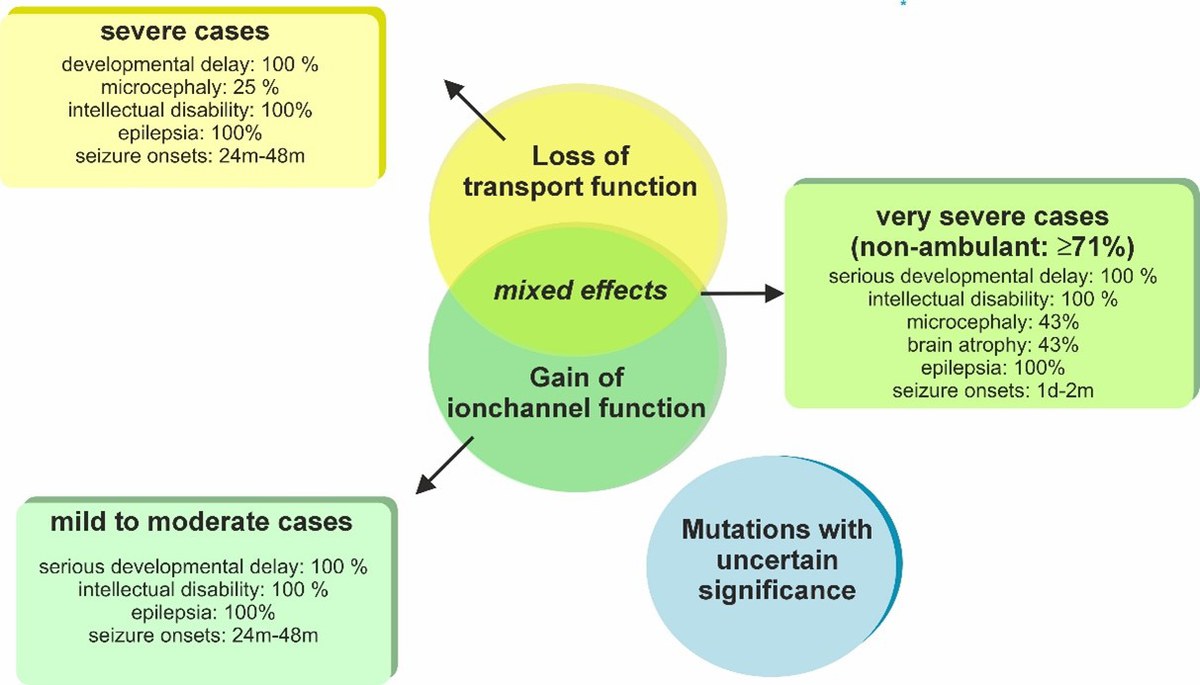
One striking finding: even a complete loss of EAAT2 function was associated with only mild symptoms in one child. In contrast, the most severe phenotypes – including drug-resistant epilepsy and pronounced brain atrophy – occurred in cases where a mutation both impaired glutamate transport and increased anion channel activity. The mildest symptoms were seen in individuals whose mutations led to only a slight increase in channel function.
The figure shows the relationship between the various functional disorders observed in genetically modified glutamate transporters and the effects on the patients affected. | Copyrights: Forschungszentrum Jülich / Peter Kovermann
Scientific and societal relevance
The study offers a clear therapeutic insight: it is not the impaired glutamate transport alone, but the increased anion channel activity of EAAT2 that appears to be the decisive pathological factor in the most severe cases. This highlights new therapeutic avenues – including drugs that selectively target and inhibit this channel activity.
The case of a mild phenotype despite the complete loss of EAAT2 function suggests that pharmacological blockade of both EAAT2 functions could represent a promising approach in the most severe forms of the disease. Overall, the findings underline the crucial importance of precise regulation of EAAT2’s anion channel function for normal brain activity.
Epileptic encephalopathies (EIEEs) are a group of neurological disorders characterised by frequent epileptic episodes in early childhood and by progressive developmental atrophy of the nervous system. The majority of cases suffer from lifelong developmental and autistic disorders. The findings from this study not only improve our understanding of the biological basis of these disorders, but may also pave the way for more personalised treatments in the future.